

Die Beschwerden zu Beginn der Erkrankung hängen davon ab, welche motorischen Nervenzellen (Motoneurone) betroffen sind. Es können sowohl Nervenzellen im Gehirn als auch im Rückenmark betroffen sein.
Die motorischen Nervenzellen im Gehirn sind die oberen Motoneurone; sie werden auch 1. Motoneuron genannt. Die Fortsätze dieser Nervenzellen reichen bis in das Rückenmark. Dort haben sie dann Kontakt zu den motorischen Nervenzellen des Rückenmarks, auch untere Motoneurone oder 2. Motoneuron genannt. Diese wiederum sind mit den Muskeln verbunden. Erste Anzeichen der ALS können bei den einzelnen Betroffenen an unterschiedlichen Stellen auftreten.
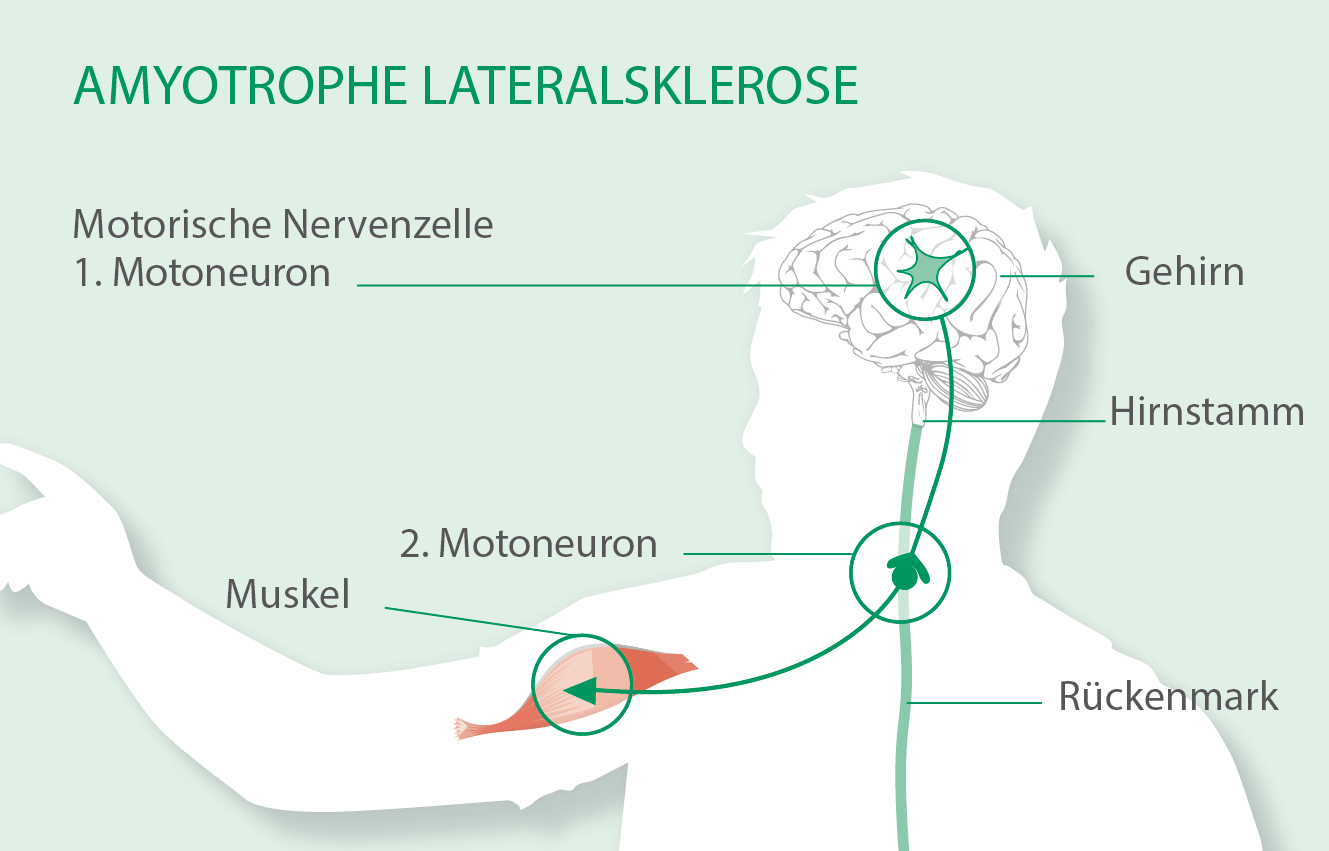
1. Motoneuron (motorische Nervenzellen im Gehirn)
2. Motoneuron (motorische Nervenzellen im Rückenmark)
Auf Symptomebene wird auch zwischen spinalem und bulbärem Beginn unterschieden.
Bei der Mehrheit der ALS-Patienten entwickelt sich die Erkrankung vom Rückenmark ausgehend. Die ersten Anzeichen treten an den Armen und Beinen auf. Bemerkbar sind häufig Muskelschwäche, Muskelzuckungen und schmerzhafte Muskelkrämpfe. Meist beginnt die Erkrankung in einer Körperregion und breitet sich von dort aus.
Bei ca. 30 % der Patienten geht die Erkrankung vom Hirnstamm aus und beginnt mit Schluck- oder Sprechstörungen. Im weiteren Krankheitsverlauf treten diese Symptome bei etwa 80 % der Erkrankten auf.


Mit dem Fortschreiten der Krankheit verstärkt sich die Schwäche der Muskulatur. Die Verringerung der Muskelmasse kann zum Beispiel an der Muskulatur zwischen Daumen und Zeigefinger sichtbar werden. Bewegungen mit den Händen, wie zum Beispiel Knöpfe schließen, werden schwieriger. Eine zunehmende Schwäche in Beinen und Füßen erschwert das Gehen.
Schreitet die Erkrankung fort, kann eine Gehstütze oder ein Rollstuhl notwendig werden. Ist die obere Körperhälfte betroffen, sind die Bewegungen, die wir mit den Schultern, Armen und Händen verrichten, beeinträchtigt. Auch das selbstständige Essen und die Körperpflege werden zunehmend schwieriger.

Bei ersten Anzeichen einer Schluckstörung sollte der behandelnde Arzt informiert werden!
Bei fast allen Patienten kommt es im Krankheitsverlauf zu Sprech-, Kau- und Schluckstörungen. Diese Störungen können so schwerwiegend sein, dass der Betroffene gar nicht mehr sprechen kann. Das Sprachverständnis bleibt jedoch vollständig erhalten.
Durch die zunehmend erschwerte Nahrungsaufnahme kommt es zu Gewichtsverlust. Um diesem entgegenzuwirken, sollte auf eine spezielle Nahrungszusammenstellung geachtet werden oder es können Ernährungshilfen zum Einsatz kommen. Im Verlauf der Erkrankung entsteht eine Schwäche der Atemmuskulatur und des Zwerchfells, dadurch kann auch die Atmung erschwert werden. Für diesen Fall gibt es verschiedene Atemhilfen, die je nach individuellem Krankheitsverlauf eingesetzt werden können. Mehr Informationen dazu erhalten Sie unter dem Menüpunkt „Tipps für den Alltag mit ALS“.
Die Diagnose der ALS erfolgt meistens durch einen Neurologen. Der Patient und die Angehörigen sollen umfassend und in Ruhe aufgeklärt werden. Oft erfolgt die Einholung einer ärztlichen Zweitmeinung.
Symptome, die auf ALS hinweisen, können auch ganz andere Ursachen haben. Daher ist es wichtig, dass ein Neurologe die Diagnose stellt. Der Neurologe führt eine klinische Untersuchung durch, bei der besonders auf Zeichen von Muskelschwund oder -schwäche bzw. unwillkürliche Muskelzuckungen oder erhöhte Muskelspannung geachtet wird. Auch das Sprechen, Schlucken und die Atemfunktion werden untersucht. Um das Ausmaß der betroffenen Muskeln genau zu erfassen oder auch um andere Erkrankungen auszuschließen, erfolgen noch Zusatzuntersuchungen, wie die Elektromyographie (EMG), Messung der Nervenleitgeschwindigkeit, bildgebende Verfahren und Laboruntersuchungen.
Die Diagnose der Erkrankung erfolgt über die typischen Symptome und eine körperliche Untersuchung. Ein typisches Merkmal der ALS ist die langsame Zunahme der Muskelschwäche oder Steifigkeit.

Meist erkranken ältere Erwachsene zwischen dem 50. und 70. Lebensjahr an ALS. Männer sind etwas häufiger betroffen als Frauen (1,6 zu 1). Der Großteil (90%) der ALS-Erkrankungen tritt „sporadisch“ auf, also nicht erblich oder vererbbar. ALS ist auch nicht ansteckend.
In Deutschland gibt es ca. 7.000 ALS-Patienten. Jedes Jahr wird die Erkrankung bei etwa 1.600 Menschen festgestellt. Das mittlere Erkrankungsalter beträgt 60 Jahre. Der Verlauf der ALS ist von Patient zu Patient sehr unterschiedlich. Die durchschnittliche Lebenserwartung von ALS-Patienten beträgt 3 bis 5 Jahre.
Bei 10-20 % der Patienten ist das Fortschreiten dagegen wesentlich langsamer. Ein Beispiel ist der britische Physiker und Astrophysiker Stephen Hawking, bei dem 1963 eine seltene Sonderform der ALS diagnostiziert wurde. Mobil blieb er durch einen Rollstuhl, zudem wurde er seit 1985 beatmet. Für die verbale Kommunikation nutzte er einen Sprachcomputer, den er allein durch die Bewegung seines Wangenmuskels und seiner Augen steuerte. Bis zu seinem Tod im März 2018 widmete er sich aktiv seiner Forschung. Auch wenn sein Krankheitsverlauf nicht die Regel ist, kann durch moderne Behandlungsoptionen und Hilfsmittel eine Lebensverlängerung bis hin zu vielen Jahren erreicht werden.
Man geht bei der ALS u.a. davon aus, dass natürlich vorkommende Eiweiße (Proteine) in den betroffenen Nervenzellen so verändert sind, dass sie sich miteinander verbinden und schließlich verklumpen. Diese Verklumpung ist so umfangreich, dass sie die Nervenzelle zerstört. Diesen Vorgang nennt man Proteinopathie. Die Ursache dafür ist noch nicht endgültig geklärt. In 5-10 % der Krankheitsfälle ist von einer vererbten ALS auszugehen (familiäre ALS).1 Die ALS ist also keine typische Erbkrankheit. Durch die Forschung sind bereits einige Gene bekannt, die mit einem erhöhten ALS-Risiko in Verbindung gebracht werden. Sollten Sie die Vermutung haben, dass in Ihrer Familie die seltene erbliche ALS vorkommt, wenden Sie sich an Ihren behandelnden Facharzt.